动脉粥样硬化是一种动脉慢性炎症性疾病,其特征是脂质成分和巨噬细胞堆积形成斑块,其中这些细胞在炎症反应中起着至关重要的作用。沿血流循环的单核细胞被募集到动脉壁,其运动受趋化因子c-c基序趋化因子配体2(CCL2)的调控,单核细胞暴露于剪切应力有助于它们更容易穿透内皮。迁移后,单核细胞分化成活化的巨噬细胞。巨噬细胞的激活在伤口愈合的组织修复和再生中起着重要作用,而活化至M1表型的巨噬细胞会分泌大量促炎细胞因子,如CCL2、IL-1β和TNF-α。因此,巨噬细胞的激活促进周围微环境调控的促炎或抗炎作用。巨噬细胞通常位于斑块破裂部位,可能受到机械刺激。然而,巨噬细胞在承受机械应力时的功能是未知的。
血流动力学剪切应力是一种机械因素,被认为是动脉粥样硬化的主要因素。为了更好地了解生物力学刺激对炎症的影响,韩国国立釜山大学医学院药学系、韩国生命工学研究院、岭南大学药学系联合团队的一项研究将人THP-1单核细胞/巨噬细胞暴露于剪切应力下,并检测炎症分子的表达。这项研究中报告了新的结果,表明生物力学刺激是激活单核细胞/巨噬细胞为促炎表型的因素之一,还确定了被认为与剪切应力诱导的炎症反应相关的膜蛋白。此外,使用人和小鼠动脉粥样硬化斑块标本研究了已识别的一种蛋白的表达,以验证结果的体内相关性。

剪切应力诱导促炎 M1 标志物的产生
为了研究剪切应力是否影响单核细胞/巨噬细胞的表型,实验利用THP-1细胞确定了剪切应力对M1/ M2标志物的时间过程影响。RT-PCR显示CCL2、IL-1β和TNF-α的转录水平在暴露于剪切应力(SS,12 dynes/cm2)12小时后开始增强,并在24小时变得明显(图1 A)。相比之下,在暴露于剪切应力24小时后,M2标记物CD163和LXRα的转录水平降低。这些结果表明,剪切应力诱导促炎M1标志物的转录。
然后通过ELISA测定了CCL2,TNF-α和IL-1β的分泌水平(图1 B)。剪切应力暴露12和24小时后,CCL2分泌增加。THP-1细胞分泌少量或几乎检测不到的TNF-α和IL-1β,但剪切应力应用12和24小时后,TNF-α 和 IL-1β分泌增加。这些结果表明,剪切应力增强了促炎分子的分泌。
这些数据表明,剪切应力在转录和蛋白质水平上增加了多个M1标记分子的单核细胞/巨噬细胞表达,极化为促炎表型。

图1 剪切应力对M1极化标记物的影响。
剪切应力对CCL2、TNF-α和IL-1β的表达是可逆的
接下来研究了M1标志物的表达是否受到剪切应力消失的影响(图1 C、D)。剪切应力处理24小时后,CCL2,TNF-α和IL-1β的转录水平增加,但在无剪切应力的正常培养条件下再培养24 小时时,转录水平显著降低(图1 C)。实验还测量了这些标志物的分泌。将剪切应力刺激的细胞分为两组:在没有剪切应力暴露的情况下再培养24小时,没有(ST)和有改变培养基(mc ST)(图1 D)。CCL2和TNF-α的分泌在剪切应力下增加,当细胞恢复到正常培养条件而没有改变培养基时继续分泌。CCL2分泌进一步增强。然而,当剪切应力细胞在正常培养条件下且改变培养基时,CCL2、TNF-α和IL-1β的分泌显著减少或降低到基础水平。这些结果表明,由剪切应力诱导的炎症分子的表达在剪切应力刺激细胞中是可逆的。
ERK 和 Src 通路抑制剂可抑制剪切应力诱导的 M1 标志物表达
由于SRC和ERK信号参与巨噬细胞极化为促炎表型,实验使用MEK抑制剂U0126评估了它们在剪切应力诱导的M1标志物表达中的作用,该抑制剂可激活ERK1/2和SRC激酶抑制剂PP2(图1 E)。暴露于剪切应力的THP-1细胞中CCL2的转录水平增加。然而,在U0126和PP2存在的情况下,这种增加开始降低。IL-1β转录水平也以类似于CCL2的模式下降,而TNF-α的转录水平没有通过两种抑制剂的处理而改变。在剪切应力下,这些结果表明,ERK和SRC信号通路参与巨噬细胞CCL2和IL-1β的表达,而不是TNF-α的表达。
剪切应力上调表层蛋白(包括 HSPs)的表达
接下来实验研究了剪切应力是否改变了细胞表层的蛋白质水平。从THP-1细胞中分离细胞表层蛋白(图2 A),然后研究了剪切应力是否会影响热休克蛋白105、热休克蛋白90、热休克蛋白70( HSP105,HSP90,HSP70)和人腺苷酸环化酶关联蛋白1( CAP1)的转录和蛋白质水平。HSP105和CAP1的转录水平没有变化,而HSP90和HSP70的转录水平增加(图2 B)。然后分析蛋白质表达(图2 C),HSP105,HSP90和HSP70的细胞水平与对照组(0 h SS)相比增加。然后测定它们在膜上的水平(图2 D),剪切应力24h后,THP-1细胞表层蛋白水平上调。在上述蛋白质中,CAP1的表达增加最低。
流式细胞术测定的暴露于剪切应力后,HSP105、HSP90 和 HSP70 的水平在 THP-1 细胞表面上调(图2 E)。当用共聚焦显微镜观察免疫染色的THP-1细胞时,观察到该蛋白质的表面表达增加(图2 F)。这些数据表明,剪切应力上调了单核细胞中HSP105,HSP90和HSP70的细胞内和表面水平。

图2 剪切应力对表层蛋白的影响。
抑制HSPs 可抑制剪切应力诱导的炎症标志物表达
实验研究了HSPs是否参与剪切应力诱导的炎症分子的表达。首先,使用两种抑制剂17-DMAG和STA-9090检查HSP90的参与。CCL2、TNF-α和IL-1β基因的转录水平在暴露于剪切应力后增加,并且在17-DMAG或STA-9090存在下显著被抑制。在17-DMAG或STA-9090存在下,剪切应力增强的CCL2,TNF-α和IL-1β的分泌被阻断。这些结果表明,HSP90的药理学抑制导致炎症反应减少。
此外还利用VER155008和knk437研究了HSP70和HSP105是否参与了促炎表型的极化。VER155008是HSP70家族的有效抑制剂,而knk437抑制多种HSPs,包括HSP105和HSP70。在VER155008和knk437存在下,剪切应力诱导的CCL2,TNF-α和IL-1β基因的转录水平受损,CCL2,TNF-α和IL-1β的分泌也受到显著抑制。其中,knk437有效抑制炎症分子的表达。这些结果表明,HSP105和HSP70抑制剂降低了炎症分子的表达。
实验观察到剪切应力后CCL2分泌增加。于是研究了分泌的CCL2是否具有功能,因为CCL2是单核细胞的有效趋化因子。剪切应力24小时后的THP-1细胞上清液中含有高水平的CCL2,增加了单核细胞的迁移。在剪切应力暴露后获得的上清液中,细胞迁移增强,而当细胞暴露于在HSP90、HSP70和HSP105抑制剂存在下的上清液时,细胞迁移降低,与CCL2分泌的结果一致。这些结果表明,HSP90、HSP70和HSP105影响剪切应力诱导的免疫细胞迁移。总的来说,这些数据表明,剪切应力后HSPs上调参与剪切应力诱导的炎症反应。
CD68 和 HSP90 在 ApoE-缺陷小鼠和人股动脉的动脉粥样硬化病变中共存与共定位
最后实验研究了HSP90在发生血流紊乱的动脉粥样硬化病变中的巨噬细胞表达。ApoE-缺陷小鼠出现动脉粥样硬化病变(图3 A)。HSP90和CD68(泛巨噬细胞标志物)免疫反应性在ApoE-缺陷小鼠的动脉粥样硬化病变中检测到,但在野生型小鼠的动脉粥样硬化病变中未检测到(图3 A)。
为了研究HSP90和CD68的共定位,使用了人类标本。在股动脉动脉粥样硬化斑块的肩部区域检测到CD68和HSP90的免疫反应性(图3 B)。当两个图像合并时,观察到两个分子的广泛共定位。一些CD68/HSP90阳性细胞位于血管内壁的管腔侧或下方,细胞容易受到剪切应力(a)。然而,在没有剪切应力的巨噬细胞缺失的区域没有检测到HSP90表达(b)。这些数据表明,体内巨噬细胞HSP90的表达在剪切应力条件下增加。

图3 免疫组织化学(IHC)分析小鼠和人源性动脉粥样硬化病变中CD68和HSP90的表达。

图4 暴露于血流的巨噬细胞介导的炎症示意图。驻留在受损动脉剥落区域的巨噬细胞受到剪切应力并分泌炎性细胞因子,包括CCL2,TNF-α和IL-1β。分泌的CCL2促进单核细胞/巨噬细胞的募集到该区域,从而增加免疫细胞的数量。受剪切应力影响的巨噬细胞也会增加HSPs的表层表达,例如HSP105,HSP90和HSP70,这是产生炎性细胞因子和趋化因子所必需的。剪切应力通过表层蛋白的上调参与巨噬细胞介导的炎症。
总之,这项研究的结果表明,剪切应力激活单核细胞/巨噬细胞,使它们上调炎症分子的表达和HSPs的细胞表层水平,如HSP70,HSP90和HSP105,这可能是主动脉血管内皮剥脱后血管炎症的治疗靶点。未来需要进一步研究HSPs在体内血管重塑中的作用,并了解机械刺激下炎症分子和HSPs表达的机制。
参考文献:Son H, Choi HS, Baek SE, Kim YH, Hur J, Han JH, Moon JH, Lee GS, Park SG, Woo CH, Eo SK, Yoon S, Kim BS, Lee D, Kim K. Shear stress induces monocyte/macrophage-mediated inflammation by upregulating cell-surface expression of heat shock proteins. Biomed Pharmacother. 2023 May;161:114566. doi: 10.1016/j.biopha.2023.114566. Epub 2023 Mar 22. PMID: 36963359.
原文链接:https://pubmed.ncbi.nlm.nih.gov/36963359/
小编旨在分享、学习、交流生物科学等领域的研究进展。如有侵权或引文不当请联系小编修正。
微信搜索公众号“Naturethink”,了解更多细胞体外仿生培养技术及应用。

新鲁汶大学的公报指出,如今抗生素耐药菌的出现给人类和医药带来了新 ...

根据“生物安全关键技术研发”重点专项评审工作安排,生物中心将于2 ...

为更好的向用户、潜在用户提供我们的产品,即日起推出如下活动:凡向 ...

2018年度国家科学技术奖提名工作已结束,国家科学技术奖励工作办 ...

据英国《自然·通讯》杂志日前发表的一篇医学论文报告,科学家发现了 ...

Naturethink祝愿伟大祖国繁荣昌盛,欣欣向荣! ...

“免疫系统在高血压中扮演了未曾预料的重要角色。”英国格拉斯哥大学 ...

“来一场中国制造的品质革命!”3月5日,李克强总理在政府工作报告 ...

我司自主研发产品,重视知识产权,已拥有多项专利证书! ...

美国侨报网近日刊文称,一项新出炉的研究警告称,即使是失眠一夜,也 ...
公司完成细胞张应变与压力综合培养仪器的研发; ...

Naturethink网站及微信内容逐步完善,敬请查阅! ...

为提高区域自主创新能力,推进区域科技创新体系建设,加大创新驱动区 ...

2018年春节将至,我司放假时间安排为:2月14日至2月21日, ...

澳大利亚和英国一项研究显示,对于几乎任何年龄段的人群而言,快走都 ...

2018年国家自然科学基金项目申请工作已开始,你准备好了吗? ...
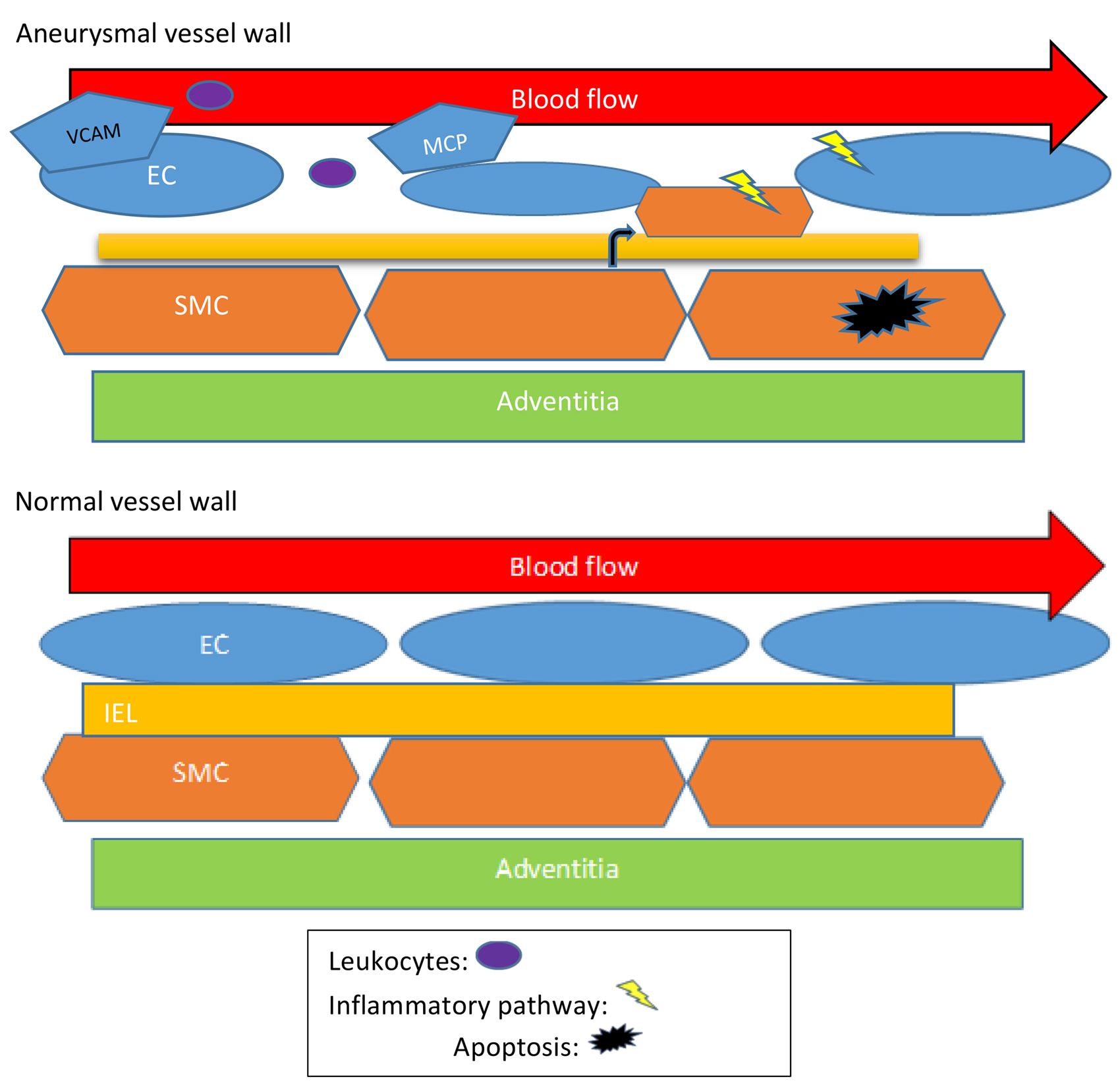
壁面剪切应力(WSS),是单位面积上由血管表面流动的液体产生的接 ...
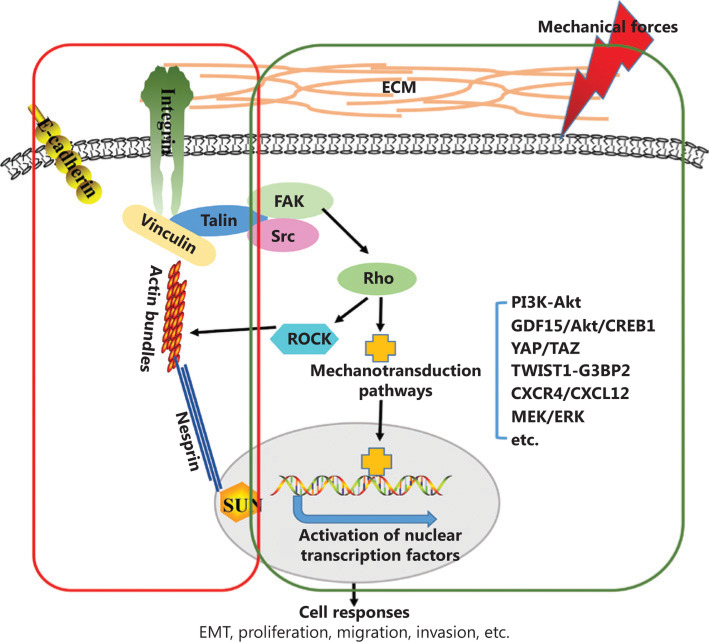
癌症在促进肿瘤表型表观遗传重编程和修饰的复杂组织微环境中发展。此 ...
